RNA-ADT: scButterfly-C¶
The following tutorial demonstrate how to use scButterfly-C variant with data augmentation using totalVI cluster labels.
scButterfly-C with totalVI cluster labels data augmentation will generate synthetic paired data by randomly paired scRNA-seq and scADT-seq of different cells with the same cluster labels. Leiden is used on the embedding space of totalVI, which is a single-cell multi-omics data joint analysis method in Python packages scvi-tools. The supplement of these generated data will provide scButterfly a better performance of translation, but take more time for training.
Note
Most of this tutorial is same as scButterfly-C for RNA and ADT data with more details of data pre-processing, model constructing, model training and evaluating. It’s prefered to see that first, because it has no different in parts mentioned above, but have more useful notes.
[1]:
import scanpy as sc
import pandas as pd
from scipy.sparse import csr_matrix
Train totalVI¶
To use totalVI augmentation, we should train a totalVI model first.
[2]:
from scvi_colab import install
install()
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scanpy as sc
import scvi
import sys
from scButterfly.calculate_cluster import *
import scipy.sparse as sp
from scButterfly.split_datasets import *
# here TotalVI need count matrixs both for RNA and ADT
adata_protein = sc.read_h5ad('CITE_BMMC_ADT.h5ad')
adata_gene = sc.read_h5ad('CITE_BMMC_RNA.h5ad')
sc.pp.highly_variable_genes(adata_gene, batch_key="batch", flavor="seurat_v3", n_top_genes=4000, subset=True)
id_list = five_fold_split_dataset(adata_gene, adata_protein, seed=19191)
train_id, validation_id, test_id = id_list[0]
adata_gene_train = adata_gene[train_id].copy()
adata_protein_train = adata_protein[train_id].copy()
adata_gene_train.obsm['protein_expression'] = adata_protein_train.to_df()
scvi.model.TOTALVI.setup_anndata(adata_gene_train, batch_key="batch", protein_expression_obsm_key="protein_expression")
model = scvi.model.TOTALVI(adata_gene_train, latent_distribution="normal", n_layers_decoder=2)
model.train()
adata_gene_train.obsm["X_totalVI"] = model.get_latent_representation()
leiden_adata = ad.AnnData(adata_gene_train.obsm["X_totalVI"])
sc.pp.neighbors(leiden_adata)
sc.tl.leiden(leiden_adata, resolution=3)
/home/atac2rna/anaconda3/envs/bt/lib/python3.9/site-packages/scvi_colab/_core.py:41: UserWarning:
Not currently in Google Colab environment.
Please run with `run_outside_colab=True` to override.
Returning with no further action.
warn(
Global seed set to 0
/home/atac2rna/anaconda3/envs/bt/lib/python3.9/site-packages/pytorch_lightning/utilities/warnings.py:53: LightningDeprecationWarning: pytorch_lightning.utilities.warnings.rank_zero_deprecation has been deprecated in v1.6 and will be removed in v1.8. Use the equivalent function from the pytorch_lightning.utilities.rank_zero module instead.
new_rank_zero_deprecation(
/home/atac2rna/anaconda3/envs/bt/lib/python3.9/site-packages/pytorch_lightning/utilities/warnings.py:58: LightningDeprecationWarning: The `pytorch_lightning.loggers.base.rank_zero_experiment` is deprecated in v1.7 and will be removed in v1.9. Please use `pytorch_lightning.loggers.logger.rank_zero_experiment` instead.
return new_rank_zero_deprecation(*args, **kwargs)
No GPU/TPU found, falling back to CPU. (Set TF_CPP_MIN_LOG_LEVEL=0 and rerun for more info.)
INFO Using column names from columns of adata.obsm['protein_expression']
INFO Computing empirical prior initialization for protein background.
GPU available: True (cuda), used: True
TPU available: False, using: 0 TPU cores
IPU available: False, using: 0 IPUs
HPU available: False, using: 0 HPUs
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0,1,2,3]
Epoch 138/138: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 138/138 [15:28<00:00, 6.45s/it, loss=1.63e+03, v_num=1]
`Trainer.fit` stopped: `max_epochs=138` reached.
Epoch 138/138: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 138/138 [15:28<00:00, 6.73s/it, loss=1.63e+03, v_num=1]
[3]:
ADT_data = sc.read_h5ad('CITE_BMMC_ADT.h5ad')
RNA_data = sc.read_h5ad('CITE_BMMC_RNA.h5ad')
ADT_data.X = csr_matrix(ADT_data.X)
RNA_data.X = csr_matrix(RNA_data.X)
[4]:
RNA_data
[4]:
AnnData object with n_obs × n_vars = 90261 × 13953
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
uns: 'dataset_id', 'genome', 'organism'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
[5]:
ADT_data
[5]:
AnnData object with n_obs × n_vars = 90261 × 134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
uns: 'dataset_id', 'genome', 'organism'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
[6]:
from scButterfly.data_processing import RNA_data_preprocessing, CLR_transform
[7]:
RNA_data = RNA_data_preprocessing(
RNA_data,
normalize_total=True,
log1p=True,
use_hvg=True,
n_top_genes=3000,
save_data=False,
file_path=None,
logging_path=None
)
ADT_data = CLR_transform(ADT_data)[0]
[INFO] RNA preprocessing: normalize size factor.
[INFO] RNA preprocessing: log transform RNA data.
[INFO] RNA preprocessing: choose top 3000 genes for following training.
[8]:
from scButterfly.split_datasets import *
id_list = five_fold_split_dataset(RNA_data, ADT_data, seed=19191)
train_id, validation_id, test_id = id_list[0]
train_id_r = train_id.copy()
train_id_a = train_id.copy()
validation_id_r = validation_id.copy()
validation_id_a = validation_id.copy()
test_id_r = test_id.copy()
test_id_a = test_id.copy()
Data augmentation with cluster labels from totalVI embeddings¶
In extensive usage, we should explicit randomly generate synthetic paired data with reference of Leiden cluster in totalVI embeddings. You could easily reporduce use the following blocks.
[9]:
copy_count = 3
random.seed(19193)
cell_type = leiden_adata.obs.leiden
for i in range(len(cell_type.cat.categories)):
cell_type_name = cell_type.cat.categories[i]
idx_temp = list(cell_type[cell_type == cell_type_name].index.astype(int))
for j in range(copy_count - 1):
random.shuffle(idx_temp)
for each in idx_temp:
train_id_r.append(train_id[each])
random.shuffle(idx_temp)
for each in idx_temp:
train_id_a.append(train_id[each])
[10]:
from scButterfly.train_model_cite import Model
import torch
import torch.nn as nn
[11]:
RNA_input_dim = len([i for i in RNA_data.var['highly_variable'] if i])
ADT_input_dim = ADT_data.X.shape[1]
R_kl_div = 1 / RNA_input_dim * 20
A_kl_div = R_kl_div
kl_div = R_kl_div + A_kl_div
[12]:
model = Model(
R_encoder_nlayer = 2,
A_encoder_nlayer = 2,
R_decoder_nlayer = 2,
A_decoder_nlayer = 2,
R_encoder_dim_list = [RNA_input_dim, 256, 128],
A_encoder_dim_list = [ADT_input_dim, 128, 128],
R_decoder_dim_list = [128, 256, RNA_input_dim],
A_decoder_dim_list = [128, 128, ADT_input_dim],
R_encoder_act_list = [nn.LeakyReLU(), nn.LeakyReLU()],
A_encoder_act_list = [nn.LeakyReLU(), nn.LeakyReLU()],
R_decoder_act_list = [nn.LeakyReLU(), nn.LeakyReLU()],
A_decoder_act_list = [nn.LeakyReLU(), nn.Identity()],
translator_embed_dim = 128,
translator_input_dim_r = 128,
translator_input_dim_a = 128,
translator_embed_act_list = [nn.LeakyReLU(), nn.LeakyReLU(), nn.LeakyReLU()],
discriminator_nlayer = 1,
discriminator_dim_list_R = [128],
discriminator_dim_list_A = [128],
discriminator_act_list = [nn.Sigmoid()],
dropout_rate = 0.1,
R_noise_rate = 0.5,
A_noise_rate = 0,
chrom_list = [],
logging_path = None,
RNA_data = RNA_data,
ATAC_data = ADT_data
)
Data augmentation will take more time for training.
[13]:
model.train(
R_encoder_lr = 0.001,
A_encoder_lr = 0.001,
R_decoder_lr = 0.001,
A_decoder_lr = 0.001,
R_translator_lr = 0.001,
A_translator_lr = 0.001,
translator_lr = 0.001,
discriminator_lr = 0.005,
R2R_pretrain_epoch = 100,
A2A_pretrain_epoch = 100,
lock_encoder_and_decoder = False,
translator_epoch = 200,
patience = 50,
batch_size = 64,
r_loss = nn.MSELoss(size_average=True),
a_loss = nn.MSELoss(size_average=True),
d_loss = nn.BCELoss(size_average=True),
loss_weight = [1, 2, 1, R_kl_div, A_kl_div, kl_div],
train_id_r = train_id_r,
train_id_a = train_id_a,
validation_id_r = validation_id_r,
validation_id_a = validation_id_a,
output_path = None,
seed = 19193,
kl_mean = True,
R_pretrain_kl_warmup = 50,
A_pretrain_kl_warmup = 50,
translation_kl_warmup = 50,
load_model = None,
logging_path = None
)
[INFO] Trainer: RNA pretraining ...
RNA pretrain: 81%|█████████████████▊ | 81/100 [41:37<10:10, 32.13s/it, train=0.0491, val=0.0516][INFO] Trainer: RNA pretraining early stop, validation loss does not improve in 50 epoches!
RNA pretrain: 81%|█████████████████▊ | 81/100 [41:37<09:45, 30.84s/it, train=0.0491, val=0.0516]
[INFO] Trainer: ADT pretraining ...
ADT pretrain: 96%|█████████████████████ | 96/100 [45:46<01:55, 28.99s/it, train=0.0578, val=0.0810][INFO] Trainer: ADT pretraining early stop, validation loss does not improve in 50 epoches!
ADT pretrain: 96%|█████████████████████ | 96/100 [45:46<01:54, 28.61s/it, train=0.0578, val=0.0810]
[INFO] Trainer: Combine training ...
Combine training: 32%|████▏ | 64/200 [2:43:51<6:18:09, 166.84s/it, train=0.5682, val=0.6287][INFO] Trainer: Combine training early stop, validation loss does not improve in 50 epoches!
Combine training: 32%|████▏ | 64/200 [2:43:51<5:48:11, 153.61s/it, train=0.5682, val=0.6287]
[14]:
A2R_predict, R2A_predict = model.test(
test_id_r = test_id_r,
test_id_a = test_id_a,
model_path = None,
load_model = False,
output_path = None,
test_cluster = False,
test_figure = False,
output_data = False,
return_predict = True
)
[INFO] Tester: get predicting ...
RNA to ATAC predicting...: 100%|██████████████████████████████████| 283/283 [00:03<00:00, 71.11it/s]
ATAC to RNA predicting...: 100%|██████████████████████████████████| 283/283 [00:04<00:00, 69.44it/s]
[INFO] Tester: calculate neighbors graph for following test ...
[15]:
from scButterfly.calculate_cluster import calculate_cluster_index
scButterfly-C with data augmentation usually get a better performance compare to scButterfly-B. However, the performance of totalVI augmentation often slightly worse than data augmentation with cell-type labels. We recommend the scButterfly-T when there has RNA_data.obs["cell_type"] and ADT_data.obs["cell_type"].
[16]:
sc.tl.tsne(A2R_predict)
sc.tl.leiden(A2R_predict)
sc.pl.tsne(A2R_predict, color=['cell_type', 'leiden'], legend_loc='on data', legend_fontsize='xx-small')

[17]:
ARI, AMI, NMI, HOM = calculate_cluster_index(A2R_predict)
print('ADT to RNA:\nARI: %.3f, \tAMI: %.3f, \tNMI: %.3f, \tHOM: %.3f' % (ARI, AMI, NMI, HOM))
ADT to RNA:
ARI: 0.276, AMI: 0.683, NMI: 0.689, HOM: 0.761, COM: 0.629
[18]:
sc.tl.tsne(R2A_predict)
sc.tl.leiden(R2A_predict)
sc.pl.tsne(R2A_predict, color=['cell_type', 'leiden'], legend_loc='on data', legend_fontsize='xx-small')
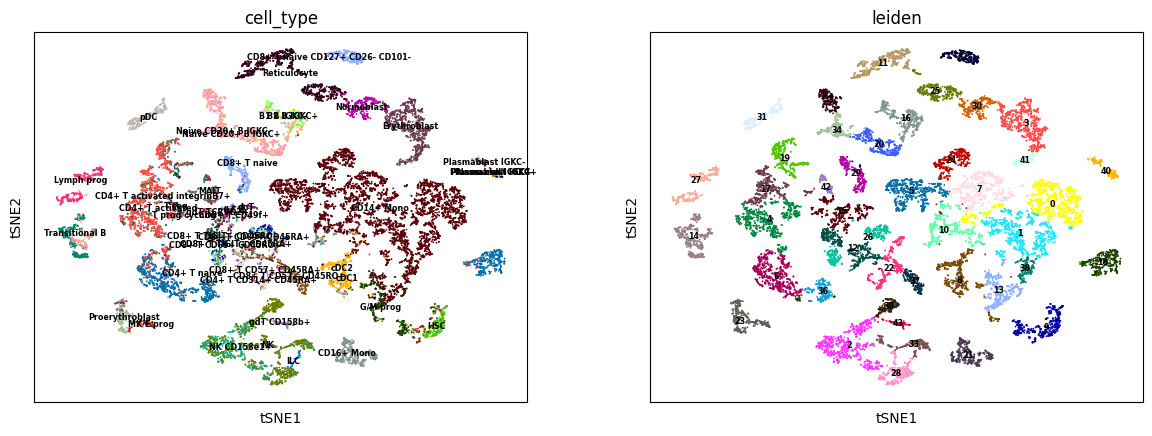
[19]:
ARI, AMI, NMI, HOM = calculate_cluster_index(R2A_predict)
print('RNA to ADT:\nARI: %.3f, \tAMI: %.3f, \tNMI: %.3f, \tHOM: %.3f' % (ARI, AMI, NMI, HOM))
RNA to ADT:
ARI: 0.321, AMI: 0.708, NMI: 0.713, HOM: 0.783, COM: 0.654
[ ]: