RNA-ATAC: unpaired data training¶
The following tutorial demonstrate how to use unpaired data to train a scButterfly model.
Get inspiration from scButterfly-T, we could randomly pair RNA profile and ATAC profile of different cell with the same cell types.
There are three part of this tutorial:
Load data and data pre-processing. This part will tell you how to load and pre-process scRNA-seq and scATAC-seq data for scButterfly model.
Construct and train a scButterfly model. This part will tell you how to generate and train a scButterfly model correctly.
Get prediction and evaluate the performance. This part will tell you how to get prediction from scButterfly model and evaluate the performance of prediction.
Note
This tutorial shows the powerful ability of versatile scButterfly framework. You could follow this tutorial to make diagnal analysis with scButterfly.
[1]:
import scanpy as sc
import pandas as pd
Load data and data pre-processing¶
Here we use the adult human kidney dataset as example. (Muto Y, et al., 2021)
[2]:
ATAC_data = sc.read_h5ad('UP_HK_ATAC.h5ad')
RNA_data = sc.read_h5ad('UP_HK_RNA.h5ad')
RNA_data.obs.index = pd.Series([str(i) for i in range(len(RNA_data.obs.index))])
ATAC_data.obs.index = pd.Series([str(i) for i in range(len(ATAC_data.obs.index))])
[3]:
RNA_data
[3]:
AnnData object with n_obs × n_vars = 19985 × 27146
obs: 'assay_ontology_term_id', 'development_stage_ontology_term_id', 'donor_uuid', 'ethnicity_ontology_term_id', 'library_uuid', 'mapped_reference_annotation', 'organism_ontology_term_id', 'sample_preservation_method', 'sample_uuid', 'suspension_type', 'suspension_uuid', 'tissue_ontology_term_id', 'is_primary_data', 'author_cell_type', 'cell_type_category', 'cell_type_ontology_term_id', 'author_cluster', 'disease_ontology_term_id', 'reported_diseases', 'sex_ontology_term_id', 'percent.mt', 'percent.rpl', 'percent.rps', 'nCount_SCT', 'nFeature_SCT', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'ethnicity', 'development_stage', 'domain', 'protocol', 'dataset', 'batch'
var: 'gene_ids', 'feature_types', 'genome', 'chrom', 'chromStart', 'chromEnd', 'name', 'score', 'strand', 'thickStart', 'thickEnd', 'itemRgb', 'blockCount', 'blockSizes', 'blockStarts', 'gene_type', 'gene_name', 'hgnc_id', 'havana_gene', 'tag', 'n_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'X_normalization', 'default_embedding', 'hvg', 'layer_descriptions', 'schema_version', 'title'
obsm: 'X_umap'
[4]:
ATAC_data
[4]:
AnnData object with n_obs × n_vars = 24205 × 99019
obs: 'assay_ontology_term_id', 'development_stage_ontology_term_id', 'donor_uuid', 'ethnicity_ontology_term_id', 'library_uuid', 'organism_ontology_term_id', 'sample_preservation_method', 'sample_uuid', 'suspension_type', 'suspension_uuid', 'tissue_ontology_term_id', 'is_primary_data', 'author_cell_type', 'cell_type_category', 'cell_type_ontology_term_id', 'author_cluster', 'disease_ontology_term_id', 'reported_diseases', 'sex_ontology_term_id', 'nCount_RNA', 'nFeature_RNA', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'ethnicity', 'development_stage', 'domain', 'protocol', 'dataset', 'batch'
var: 'chrom', 'chromStart', 'chromEnd', 'genome', 'n_counts'
uns: 'X_normalization', 'default_embedding', 'layer_descriptions', 'schema_version', 'title'
obsm: 'X_umap'
First, we pre-process data using RNA_data_preprocessing and ATAC_data_preprocessing in scButterfly.data_processing.
[5]:
from scButterfly.data_processing import RNA_data_preprocessing, ATAC_data_preprocessing
[6]:
RNA_data = RNA_data_preprocessing(
RNA_data,
normalize_total=True,
log1p=True,
use_hvg=True,
n_top_genes=3000,
save_data=False,
file_path=None,
logging_path=None
)
ATAC_data = ATAC_data_preprocessing(
ATAC_data,
binary_data=True,
filter_features=True,
fpeaks=0.005,
tfidf=True,
normalize=True,
save_data=False,
file_path=None,
logging_path=None
)[0]
[INFO] RNA preprocessing: normalize size factor.
[INFO] RNA preprocessing: log transform RNA data.
[INFO] RNA preprocessing: choose top 3000 genes for following training.
[INFO] ATAC preprocessing: binarizing data.
[INFO] ATAC preprocessing: filter out peaks appear lower than 0.5% cells.
[INFO] ATAC preprocessing: TF-IDF transformation.
[INFO] ATAC preprocessing: normalizing data.
Here we sample some synthetic paired RNA and ATAC of different cells in the same cell types. You could use unpaired_split_dataset in scButterfly.split_datasets to reproduce the process of sampling same with scButterfly manuscript.
[7]:
from scButterfly.split_datasets import *
id_list = unpaired_split_dataset(RNA_data, ATAC_data)
train_id_r, train_id_a, validation_id_r, validation_id_a, test_id_r, test_id_a = id_list[0]
Construct and train a scButterfly model¶
Calculate the counts of peaks in each chromosomes.
[8]:
ATAC_data.var.chrom
[8]:
peaks
chr1:826622-827992 chr1
chr1:835447-835975 chr1
chr1:869609-870367 chr1
chr1:876650-877672 chr1
chr1:903810-907253 chr1
...
chrX:155264099-155264764 chrX
chrX:155612050-155613299 chrX
chrX:155767079-155768180 chrX
chrX:155820008-155820575 chrX
chrX:155880588-155881957 chrX
Name: chrom, Length: 98780, dtype: category
Categories (23, object): ['chr1', 'chr2', 'chr3', 'chr4', ..., 'chr20', 'chr21', 'chr22', 'chrX']
[9]:
chrom_list = []
last_one = ''
for i in range(len(ATAC_data.var.chrom)):
temp = ATAC_data.var.chrom[i]
if temp[0 : 3] == 'chr':
if not temp == last_one:
chrom_list.append(1)
last_one = temp
else:
chrom_list[-1] += 1
else:
chrom_list[-1] += 1
print(chrom_list, end="")
[9239, 4854, 5125, 4783, 2765, 3240, 3198, 3357, 4105, 2316, 2999, 7909, 2492, 1082, 1955, 6678, 5157, 5448, 5938, 4946, 4392, 4461, 2341]
[10]:
sum(chrom_list)
[10]:
98780
We could load scButterfly model for scRNA-seq and scATAC-seq from scButterfly.train_model
Warning
We propose you to ensure that the settings of parameters for pre-processing, construct model and train model are same with here, while feel free to decide path for logging and model output.
[11]:
from scButterfly.train_model import Model
import torch
import torch.nn as nn
[12]:
RNA_input_dim = len([i for i in RNA_data.var['highly_variable'] if i])
ATAC_input_dim = ATAC_data.X.shape[1]
R_kl_div = 1 / RNA_input_dim * 20
A_kl_div = 1 / ATAC_input_dim * 20
kl_div = R_kl_div + A_kl_div
[13]:
model = Model(
R_encoder_nlayer = 2,
A_encoder_nlayer = 2,
R_decoder_nlayer = 2,
A_decoder_nlayer = 2,
R_encoder_dim_list = [RNA_input_dim, 256, 128],
A_encoder_dim_list = [ATAC_input_dim, 32 * len(chrom_list), 128],
R_decoder_dim_list = [128, 256, RNA_input_dim],
A_decoder_dim_list = [128, 32 * len(chrom_list), ATAC_input_dim],
R_encoder_act_list = [nn.LeakyReLU(), nn.LeakyReLU()],
A_encoder_act_list = [nn.LeakyReLU(), nn.LeakyReLU()],
R_decoder_act_list = [nn.LeakyReLU(), nn.LeakyReLU()],
A_decoder_act_list = [nn.LeakyReLU(), nn.Sigmoid()],
translator_embed_dim = 128,
translator_input_dim_r = 128,
translator_input_dim_a = 128,
translator_embed_act_list = [nn.LeakyReLU(), nn.LeakyReLU(), nn.LeakyReLU()],
discriminator_nlayer = 1,
discriminator_dim_list_R = [128],
discriminator_dim_list_A = [128],
discriminator_act_list = [nn.Sigmoid()],
dropout_rate = 0.1,
R_noise_rate = 0.5,
A_noise_rate = 0.3,
chrom_list = chrom_list,
logging_path = None,
RNA_data = RNA_data,
ATAC_data = ATAC_data
)
Train a scButterfly-T model.
[14]:
model.train(
R_encoder_lr = 0.001,
A_encoder_lr = 0.001,
R_decoder_lr = 0.001,
A_decoder_lr = 0.001,
R_translator_lr = 0.001,
A_translator_lr = 0.001,
translator_lr = 0.001,
discriminator_lr = 0.005,
R2R_pretrain_epoch = 100,
A2A_pretrain_epoch = 100,
lock_encoder_and_decoder = False,
translator_epoch = 200,
patience = 50,
batch_size = 64,
r_loss = nn.MSELoss(size_average=True),
a_loss = nn.BCELoss(size_average=True),
d_loss = nn.BCELoss(size_average=True),
loss_weight = [1, 2, 1, R_kl_div, A_kl_div, kl_div],
train_id_r = train_id_r,
train_id_a = train_id_a,
validation_id_r = validation_id_r,
validation_id_a = validation_id_a,
output_path = None,
seed = 19193,
kl_mean = True,
R_pretrain_kl_warmup = 50,
A_pretrain_kl_warmup = 50,
translation_kl_warmup = 50,
load_model = None,
logging_path = None
)
[INFO] Trainer: RNA pretraining ...
RNA pretrain: 100%|█████████████████████| 100/100 [23:35<00:00, 14.15s/it, train=0.0496, val=0.0319]
[INFO] Trainer: ATAC pretraining ...
ATAC pretrain: 100%|████████████████████| 100/100 [25:04<00:00, 15.04s/it, train=0.0393, val=0.0382]
[INFO] Trainer: Combine training ...
Combine training: 33%|█████▉ | 66/200 [23:17<47:20, 21.20s/it, train=0.2605, val=0.2188][INFO] Trainer: Combine training early stop, validation loss does not improve in 50 epoches!
Combine training: 33%|█████▉ | 66/200 [23:17<47:17, 21.18s/it, train=0.2605, val=0.2188]
Get prediction and evaluate the performance¶
You could get cross-modal predictions using model.test using return_predict=True. We also provided more information metrics in this function, see in API.
[15]:
A2R_predict, R2A_predict = model.test(
test_id_r = test_id_r,
test_id_a = test_id_a,
model_path = None,
load_model = False,
output_path = None,
test_cluster = False,
test_figure = False,
output_data = False,
return_predict = True
)
[INFO] Tester: get predicting ...
RNA to ATAC predicting...: 100%|████████████████████████████████████| 86/86 [00:03<00:00, 22.03it/s]
ATAC to RNA predicting...: 100%|██████████████████████████████████| 105/105 [00:06<00:00, 15.42it/s]
[INFO] Tester: calculate neighbors graph for following test ...
Here we draw the t-SNE embeddings and measure the ARI, AMI, NMI, and HOM.
[16]:
from scButterfly.calculate_cluster import calculate_cluster_index
[17]:
sc.tl.tsne(A2R_predict)
sc.tl.leiden(A2R_predict)
sc.pl.tsne(A2R_predict, color=['cell_type', 'leiden'], legend_loc='on data', legend_fontsize='small')
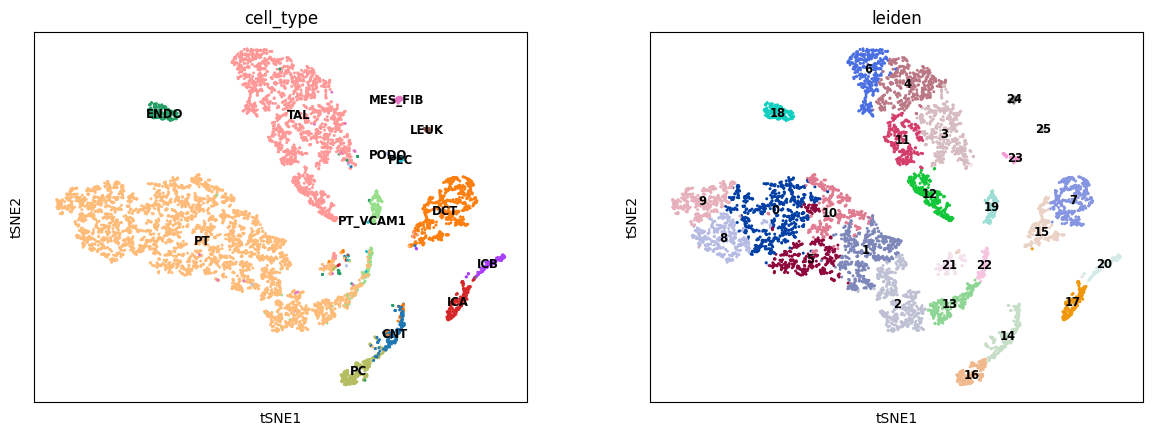
[18]:
ARI, AMI, NMI, HOM = calculate_cluster_index(A2R_predict)
print('ATAC to RNA:\nARI: %.3f, \tAMI: %.3f, \tNMI: %.3f, \tHOM: %.3f' % (ARI, AMI, NMI, HOM))
ATAC to RNA:
ARI: 0.208, AMI: 0.621, NMI: 0.625, HOM: 0.916, COM: 0.474
[19]:
sc.tl.tsne(R2A_predict)
sc.tl.leiden(R2A_predict)
sc.pl.tsne(R2A_predict, color=['cell_type', 'leiden'], legend_loc='on data', legend_fontsize='small')
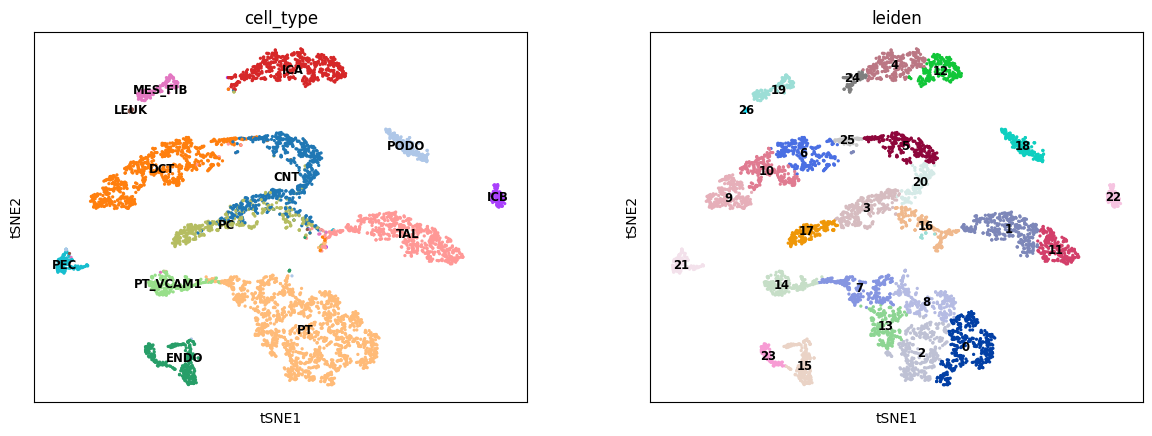
[20]:
ARI, AMI, NMI, HOM = calculate_cluster_index(R2A_predict)
print('RNA to ATAC:\nARI: %.3f, \tAMI: %.3f, \tNMI: %.3f, \tHOM: %.3f' % (ARI, AMI, NMI, HOM))
RNA to ATAC:
ARI: 0.411, AMI: 0.755, NMI: 0.758, HOM: 0.925, COM: 0.642