RNA-ADT: scButterfly-T¶
The following tutorial demonstrate how to use scButterfly-T variant with data augmentation using cell-type labels.
scButterfly-T with cell-type labels data augmentation will generate synthetic paired data by randomly paired scRNA-seq and scADT-seq of different cells with the same cell type. The supplement of these generated data will provide scButterfly-T a better performance of translation, but take more time for training.
Note
Most of this tutorial is same as scButterfly-B for RNA and ADT data with more details of data pre-processing, model constructing, model training and evaluating. It’s prefered to see that first, because it has no different in parts mentioned above, but have more useful notes.
[1]:
import scanpy as sc
import pandas as pd
from scipy.sparse import csr_matrix
import random
[2]:
ADT_data = sc.read_h5ad('CITE_BMMC_ADT.h5ad')
RNA_data = sc.read_h5ad('CITE_BMMC_RNA.h5ad')
ADT_data.X = csr_matrix(ADT_data.X)
RNA_data.X = csr_matrix(RNA_data.X)
[3]:
RNA_data
[3]:
AnnData object with n_obs × n_vars = 90261 × 13953
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
uns: 'dataset_id', 'genome', 'organism'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
[4]:
ADT_data
[4]:
AnnData object with n_obs × n_vars = 90261 × 134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
uns: 'dataset_id', 'genome', 'organism'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
[5]:
from scButterfly.data_processing import RNA_data_preprocessing, CLR_transform
[6]:
RNA_data = RNA_data_preprocessing(
RNA_data,
normalize_total=True,
log1p=True,
use_hvg=True,
n_top_genes=3000,
save_data=False,
file_path=None,
logging_path=None
)
ADT_data = CLR_transform(ADT_data)[0]
[INFO] RNA preprocessing: normalize size factor.
[INFO] RNA preprocessing: log transform RNA data.
[INFO] RNA preprocessing: choose top 3000 genes for following training.
[7]:
from scButterfly.split_datasets import *
id_list = five_fold_split_dataset(RNA_data, ADT_data, seed=19191)
train_id, validation_id, test_id = id_list[0]
train_id_r = train_id.copy()
train_id_a = train_id.copy()
validation_id_r = validation_id.copy()
validation_id_a = validation_id.copy()
test_id_r = test_id.copy()
test_id_a = test_id.copy()
Data augmentation with cell-type labels¶
In extensive usage, we should explicit randomly generate synthetic paired data with reference of RNA_data.obs["cell_type"] or ADT_data.obs["cell_type"]. You could easily reporduce use the following blocks.
[8]:
copy_count = 3
random.seed(19193)
ADT_data.obs.index = [str(i) for i in range(len(ADT_data.obs.index))]
cell_type = ADT_data.obs.cell_type.iloc[train_id]
for i in range(len(cell_type.cat.categories)):
cell_type_name = cell_type.cat.categories[i]
idx_temp = list(cell_type[cell_type == cell_type_name].index.astype(int))
for j in range(copy_count - 1):
random.shuffle(idx_temp)
train_id_r.extend(idx_temp)
random.shuffle(idx_temp)
train_id_a.extend(idx_temp)
Warning
To use data augmentation with cell-type labels, you should ensure that there has cell_type label in RNA_data.obs and ADT_data.obs. If you don’t have information about cell types, we suggest use scButterfly-C as substitution.
[9]:
from scButterfly.train_model_cite import Model
import torch
import torch.nn as nn
[10]:
RNA_input_dim = len([i for i in RNA_data.var['highly_variable'] if i])
ADT_input_dim = ADT_data.X.shape[1]
R_kl_div = 1 / RNA_input_dim * 20
A_kl_div = R_kl_div
kl_div = R_kl_div + A_kl_div
[11]:
model = Model(
R_encoder_nlayer = 2,
A_encoder_nlayer = 2,
R_decoder_nlayer = 2,
A_decoder_nlayer = 2,
R_encoder_dim_list = [RNA_input_dim, 256, 128],
A_encoder_dim_list = [ADT_input_dim, 128, 128],
R_decoder_dim_list = [128, 256, RNA_input_dim],
A_decoder_dim_list = [128, 128, ADT_input_dim],
R_encoder_act_list = [nn.LeakyReLU(), nn.LeakyReLU()],
A_encoder_act_list = [nn.LeakyReLU(), nn.LeakyReLU()],
R_decoder_act_list = [nn.LeakyReLU(), nn.LeakyReLU()],
A_decoder_act_list = [nn.LeakyReLU(), nn.Identity()],
translator_embed_dim = 128,
translator_input_dim_r = 128,
translator_input_dim_a = 128,
translator_embed_act_list = [nn.LeakyReLU(), nn.LeakyReLU(), nn.LeakyReLU()],
discriminator_nlayer = 1,
discriminator_dim_list_R = [128],
discriminator_dim_list_A = [128],
discriminator_act_list = [nn.Sigmoid()],
dropout_rate = 0.1,
R_noise_rate = 0.5,
A_noise_rate = 0,
chrom_list = [],
logging_path = None,
RNA_data = RNA_data,
ATAC_data = ADT_data
)
Data augmentation will take more time for training.
[12]:
model.train(
R_encoder_lr = 0.001,
A_encoder_lr = 0.001,
R_decoder_lr = 0.001,
A_decoder_lr = 0.001,
R_translator_lr = 0.001,
A_translator_lr = 0.001,
translator_lr = 0.001,
discriminator_lr = 0.005,
R2R_pretrain_epoch = 100,
A2A_pretrain_epoch = 100,
lock_encoder_and_decoder = False,
translator_epoch = 200,
patience = 50,
batch_size = 64,
r_loss = nn.MSELoss(size_average=True),
a_loss = nn.MSELoss(size_average=True),
d_loss = nn.BCELoss(size_average=True),
loss_weight = [1, 2, 1, R_kl_div, A_kl_div, kl_div],
train_id_r = train_id_r,
train_id_a = train_id_a,
validation_id_r = validation_id_r,
validation_id_a = validation_id_a,
output_path = None,
seed = 19193,
kl_mean = True,
R_pretrain_kl_warmup = 50,
A_pretrain_kl_warmup = 50,
translation_kl_warmup = 50,
load_model = None,
logging_path = None
)
[INFO] Trainer: RNA pretraining ...
RNA pretrain: 72%|███████████████▊ | 72/100 [32:56<11:57, 25.63s/it, train=0.0492, val=0.0517][INFO] Trainer: RNA pretraining early stop, validation loss does not improve in 50 epoches!
RNA pretrain: 72%|███████████████▊ | 72/100 [32:56<12:48, 27.45s/it, train=0.0492, val=0.0517]
[INFO] Trainer: ADT pretraining ...
ADT pretrain: 100%|█████████████████████| 100/100 [46:14<00:00, 27.74s/it, train=0.0552, val=0.0790]
[INFO] Trainer: Combine training ...
Combine training: 29%|███▊ | 58/200 [2:12:26<6:02:01, 152.97s/it, train=0.5706, val=0.6340][INFO] Trainer: Combine training early stop, validation loss does not improve in 50 epoches!
Combine training: 29%|███▊ | 58/200 [2:12:26<5:24:15, 137.01s/it, train=0.5706, val=0.6340]
[13]:
A2R_predict, R2A_predict = model.test(
test_id_r = test_id_r,
test_id_a = test_id_a,
model_path = None,
load_model = False,
output_path = None,
test_cluster = False,
test_figure = False,
output_data = False,
return_predict = True
)
[INFO] Tester: get predicting ...
RNA to ATAC predicting...: 100%|██████████████████████████████████| 283/283 [00:04<00:00, 64.58it/s]
ATAC to RNA predicting...: 100%|██████████████████████████████████| 283/283 [00:04<00:00, 66.88it/s]
[INFO] Tester: calculate neighbors graph for following test ...
[14]:
from scButterfly.calculate_cluster import calculate_cluster_index
scButterfly-T usually get a better performance compare to scButterfly-B and scButterfly-C.
[15]:
sc.tl.tsne(A2R_predict)
sc.tl.leiden(A2R_predict)
sc.pl.tsne(A2R_predict, color=['cell_type', 'leiden'], legend_loc='on data', legend_fontsize='xx-small')
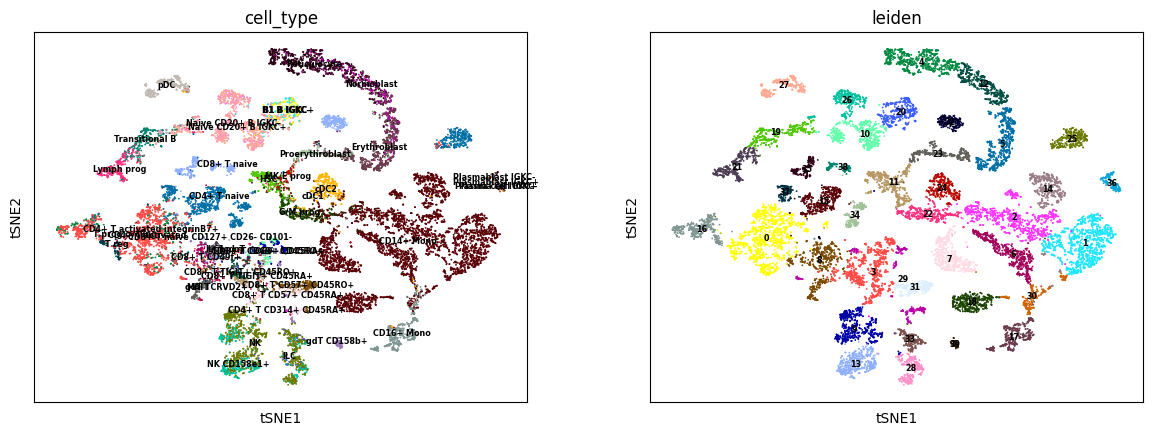
[16]:
ARI, AMI, NMI, HOM = calculate_cluster_index(A2R_predict)
print('ADT to RNA:\nARI: %.3f, \tAMI: %.3f, \tNMI: %.3f, \tHOM: %.3f' % (ARI, AMI, NMI, HOM))
ADT to RNA:
ARI: 0.335, AMI: 0.704, NMI: 0.708, HOM: 0.764, COM: 0.660
[17]:
sc.tl.tsne(R2A_predict)
sc.tl.leiden(R2A_predict)
sc.pl.tsne(R2A_predict, color=['cell_type', 'leiden'], legend_loc='on data', legend_fontsize='xx-small')

[18]:
ARI, AMI, NMI, HOM = calculate_cluster_index(R2A_predict)
print('RNA to ADT:\nARI: %.3f, \tAMI: %.3f, \tNMI: %.3f, \tHOM: %.3f' % (ARI, AMI, NMI, HOM))
RNA to ADT:
ARI: 0.346, AMI: 0.722, NMI: 0.726, HOM: 0.792, COM: 0.671